Metagenomics 101
Complex communities of microorganisms (microbiomes) play important roles in many ecosystems, such as oceans and soil, where they are the main drivers of most biogeochemical cycles. Moreover, the microbiome inhabiting the human gastrointestinal tract have lately been shown to play important roles for maintaining the health of its host. Since microbial communities are often comprised of thousands of species, most of which are very difficult to culture, studying them requires advanced technologies. Sequencing of the genomes of organisms, from viruses and bacteria to plants and animals, has fundamentally increased our understanding of different life forms. Recently, DNA sequencing has also proved to be extremely powerful for studying the genomes within microbiomes without prior cultivation - a method called metagenomics. Metagenomic data reveals the species composition of a microbial community as well as its functional potential by decoding its functional genes. Metagenomics is however a complex and technically demanding field with ample room for improvements in both laboratory and computational (bioinformatics) methodology.

Metagenomics Outline
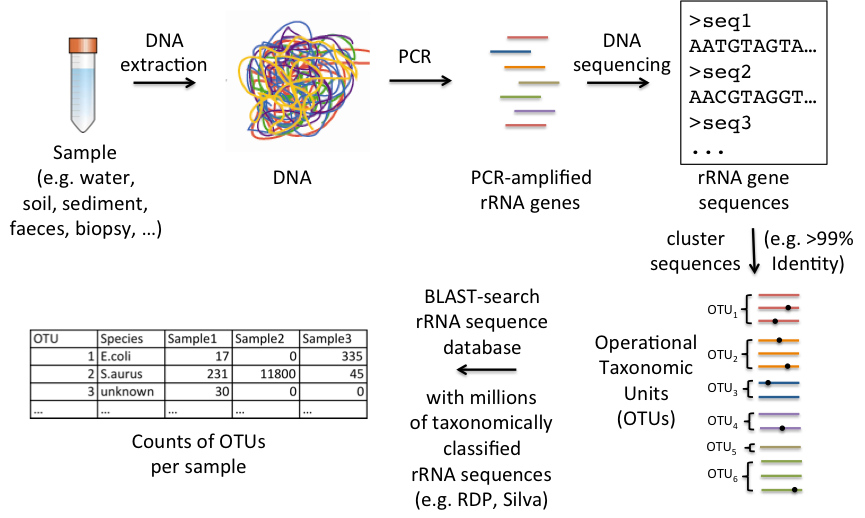
A cheaper alternative to shotgun metagenomics is the sequencing of PCR amplified taxonomic marker genes, such as rRNA genes. This doesn’t give any direct insights into the functional capabilities of the community but reveals its taxonomic composition. We typically sequence hundreds of samples at the same time and by using sample specific barcode sequences we can deduce from which sample the different sequence reads derive from. We typically get ~100,000 sequences per sample which allows us to quantify the different species with accuracy. Follow this link for our lab and bioinformatics protocols for amplicon sequencing.